
Human genetic variation database, a reference database of genetic variations in the Japanese population



 The tendency was similar to European Americans rather than African Americans (Supplementary Figure S9 in Tennessen et al.5).
The tendency was similar to European Americans rather than African Americans (Supplementary Figure S9 in Tennessen et al.5).
FDA論文も同様の立場に立っている
Koichiro Higasa, 他
nature 25 February 2016
日本人対象の全エクソンシーケンス論文
要約から
Here, we have collected exomic genetic variation from 1208 Japanese
individuals through a collaborative effort, and aggregated the data into
a prevailing catalog. In total, we identified 156 622 previously
unreported variants.
(サンプル日本人1人当たりエキソン領域で約130の新規SNV)
The allele frequencies for the majority (88.8%) were lower than 0.5% in
allele frequency and predicted to be functionally deleterious.
(その約9割の頻度は、0.5%より低い。)
In addition, we have constructed a Japanese-specific major allele
reference genome by which the number of unique mapping of the short
reads in our data has increased 0.045% on average.
前置きから
In this study, we collected exomic sequencing data of 1208 Japanese
individuals from five institutes and a data set of common variants
determined by Illumina’s BeadArray technology from 3248 individuals of
Japanese cohorts.
(5機関からの1208のエクソン領域サンプル、5機関は、捕捉表1によれば、東大(373)、京大(300)、東北大(38)、横浜市立大(429)、National Research Institute for Child Health and Development(68) の5つで、カッコ書きは、表2に示されたサンプル数
We centralized these data sets into a newly developed public database—human genetic variation database (HGVD).
手法から
サンプルから採取したのは、唾液ではなく血液を使っている
Each institute has ensured that all of the subjects have no clinical record associated with major diseases.
一応は、健常者サンプルである。(大学病院で検査用に採取した血液を本人同意の上、カルテで、病歴のない者を選んで使ったと思われる。はっきり書いてない)
Identification of novel variations and functional prediction of genetic variations
Variations were categorized as novel if they were not registered in the dbSNP (Build 137),12 the 1000 Genomes Project (November 2010 data release),13 10 personal genomes (version 1.04)14 or the NHLBI GO Exome Sequencing Project (ESP6500SI).5
(novelかどうかの判定は、上の4つとの突合による)
Allele frequency spectrum
Under the assumption of neutral evolution at equilibrium, the expected number of sites at which the new nucleotide is present x times in the sample is given by 4Nμ/x, where N and μ are the effective population size and mutation rate, respectively.20
To compute the expected spectrum, 4Nμ is estimated from the observed segregating sites according to Watterson’s formula.21
In order to make comparisons to neutral without the effect of
misidentification of ancestral states for these sites, the folded allele
frequency spectra by minor allele count were projected. Only autosomal
genes were included in calculating the allele frequency spectra.
以降飛ばした
結果から
Japanese genetic variation database
The HGVD is a web-accessible resource of genetic variations of the Japanese population. Currently, the database contains 287 588 single-nucleotide variations identified by whole-exome sequencing of 1208 individuals and 1 794 196 variants by genome scan of 3248 individuals with no record of major diseases.日本にhuman genetic variation database (HGVD). となずけた公開データベースがあるそうだ
Exome sequencing
We sequenced 1208 healthy Japanese individuals. A total of 12.9 terabases of DNA sequence were generated
Allele frequency spectrum and functional impact of Japanese genetic variations
We identified 287 588 single nucleotide variants from the filtered data
set of which 130 966 (45.5%) variants were found in the public database.
①287 588でサンプル1名平均で238
②新しく見つけたのは、差の156692で、サンプル1名平均で130
Although the minor allele frequencies of the majority of the newly
identified variants (139 096 or 88.8%) were smaller than 0.5%, the other
17 526 variants were found to be having minor allele frequency of
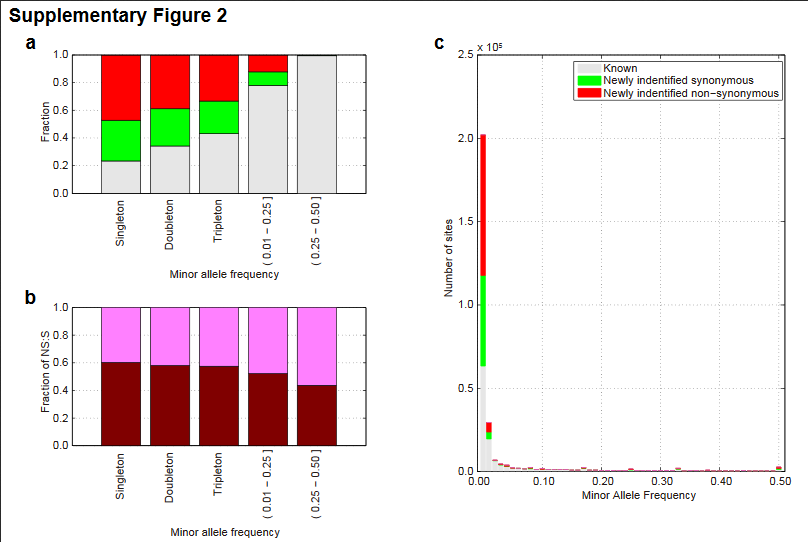
greater than 0.5% (Figure 1a and Supplementary Figure 2).

上の図の色分けは次のとおり
Minor Allele Frequency; MAF
上の図の色分けは次のとおり
Frequency and functional spectrum of variations in the Japanese. (a)
The proportion of newly identified non-synonymous, synonymous
substitution and known variations in coding regions are indicated in
red, green and gray bars, respectively. Known variations were defined as
those that were previously reported in the public databases.
MAFは、下記の略語
下が捕捉図2cで分かり易い。Cで見ると、当たり前だが、サンプル中で、日本人が共有する非同義変異は、ほとんどない。事実上、約1200名のサンプルで、1名又は2名にしかない。この一人毎の非同義変異が、アレルギー反応やいろんな薬の効き目の相違の原因なのだろう。ただし、この論文で発見されたものに限定されるが、全体の傾向とみていい。
グラフの元になる表を出しておくべきだと思う
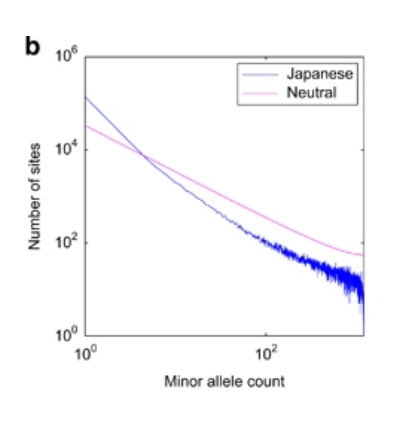
The allele frequency spectrum of the Japanese population showed an
excess of rare variations in comparison with the frequency spectrum
predicted under the neutral equilibrium model (Figure 1b).
これに対して、朝鮮人は、
①FDA論文(35名)では、
In contrast with the SNVs common to other populations in HapMap and 1KGP,
the Korean only SNVs had high percentages of non-silent variants,
emphasizing the unique roles of these Korean only SNVs in the Korean
population.Specifically,
we identified 8,361 non-synonymous Korean only SNVs, of which 58 SNVs
existed in all 35 Korean individuals.サンプル35名全員が共有している非同義変異が存在する。従って、FDAは、個別化医療で朝鮮人を別扱いするべきだとしたのだ。
非同義変異を集団内で共有しているか?=アレル頻度が高いかどうか?が重要なのだ。日本人はほぼ共有しておらず、個体差であるが、彼らは共有しており、集団形質・特性とみていい。何でこんな変なことが生じた????
②FDAと全く同じ元データを使用し、サンプル数が50名と、FDA論文より15名だけ多い韓国人論文では、サンプル平均で、非同義変異が同義変異の約半分という驚愕のデータがでいている。エクソン領域ではなく、全てでこの数値だ!

③韓国人の書いたほぼ同じ手法の論文では、サンプル1055で、同じようにエキソン領域だけ調べ、置換だけみても、非同義変異が同義変異の2倍、挿入と欠失のindelを含めれば、もっと多い。
日本人では、同じようにトータルの数値を出せば、当たり前だが、確実に、同義変異の方が非同義変異よりも多い。

To evaluate the functional impact of variations found in Japanese, we
used four measures; categories of synonymous and non-synonymous (NS:S),
PolyPhen-2,16 SIFT17 and PhyloP.18
In accordance with previous reports,5, 32 we observed an increased fraction of deleterious non-synonymous variations with lower minor allele frequencies (Figures 1c and d), suggesting that such variations arose recently enough to escape from purges of negative selection pressures.
(なるほど!韓国人は異常なまでに非同義変異が多いが、recentlyに生じたのかも、しかし、どれくらいがrecentlyかが全然書いてない。多分過去数百年としていい)
Signatures of natural selection in the Japanese genome
High frequencies of derived alleles (rs1800414 and rs885479) of these
regions were observed in Japanese (78.97 and 56.40%) compared with
European Americans (4.82 and 0.03%) and African Americans (1.53 and
0.09%).
The results support the signatures of recent positive selection, which
were observed as significant extensions of haplotype homozygosity of
these gene regions.35, 36
positive selectionについてののもであるが興味ない。私が知りたいのはnegativeだ
Construction of Japanese major-allele reference sequence
It has been shown that the ethnicity-specific major-allele reference
sequence could improve genotyping accuracy for disease-associated
variant loci.38
FDA論文も同様の立場に立っている
To apply this strategy for Japanese genomes, we substituted 816 991
positions of single nucleotide at the reference genome by the
Japanese-specific major allele.
By using 100-bases paired-end reads of
independent exomic resequencing data, we were able to uniquely map
0.045% more reads to the Japanese-specific major allele reference
sequence genome than to the NCBI reference sequence (Figure 4), due to the reduction in inconsistency of alignments (Supplementary Figure 4 and Supplementary Table 5).
Discussionより
大したことは書いてない

コメント